Projects
Prediction of reorganization energy using machine learning
Publication link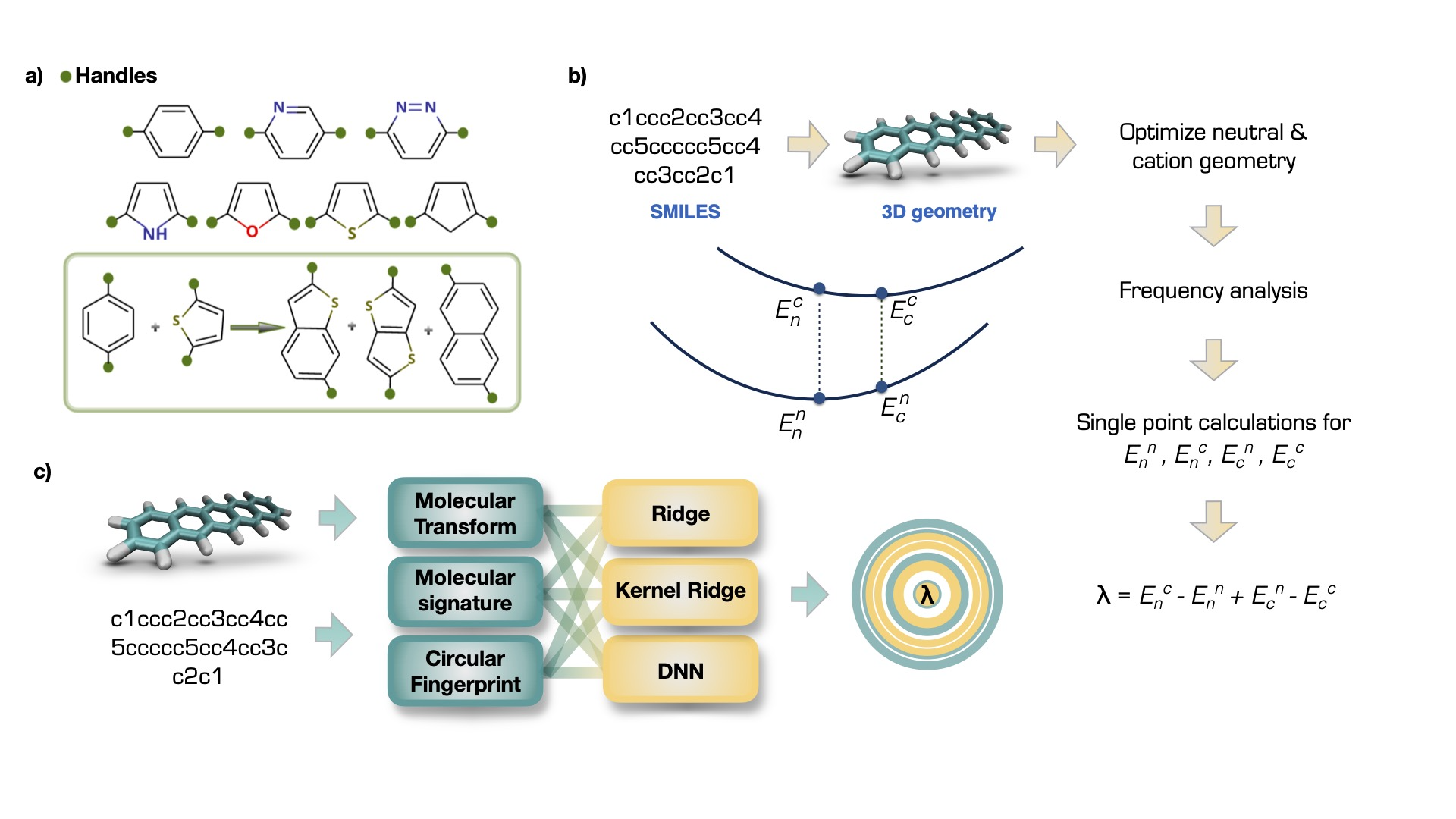
Organic semiconductors are an important class of (opto)electronic materials, with a wide range of potential applications from photovoltaics to artificial nerves. The goal of this project was to enable large-scale screening for high-performance organic semiconductors by rapid prediction of reorganization energy (RE) using machine-learning methods. It was featured on the cover of ACS magazine JPCA.
The project involved creation of a dataset using a SMILES and SMARTS based combinatorial molecule generation (a), calculation of the reorganization energy with DFT methods (b), and prediction of the reorganization energy with Ridge Regression, Kernel Ridge Regression and Deep Neural Networkds (c). (lambda represents the target reorganization energy value.)
We found that deep neural networks outperform the other methods and can predict the RE with a coefficient of determination of 0.92 and root-mean-square error of ∼12 meV. This study showed that the REs of organic semiconductor molecules can be predicted from the molecular structures with high accuracy.
An ETL, analysis and visualization project with credit card data
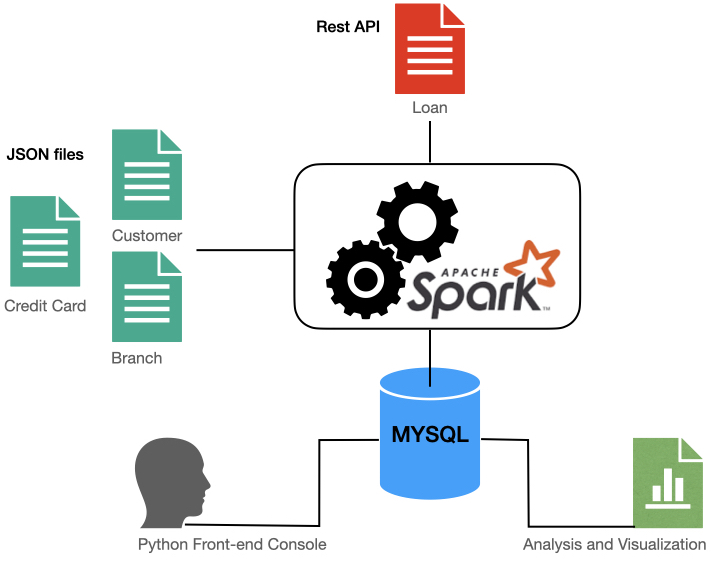
I recently finished a data engineering bootcamp where I created an ETL pipeline and application front-end with Python as the capstone project.
It was a fun project bringing together tools such as Apache Spark, Pandas, MySQL, Plotly and Tableau.
Theoretical Characterization of Charge Transport in Zinc-Chlorin Nanotubes
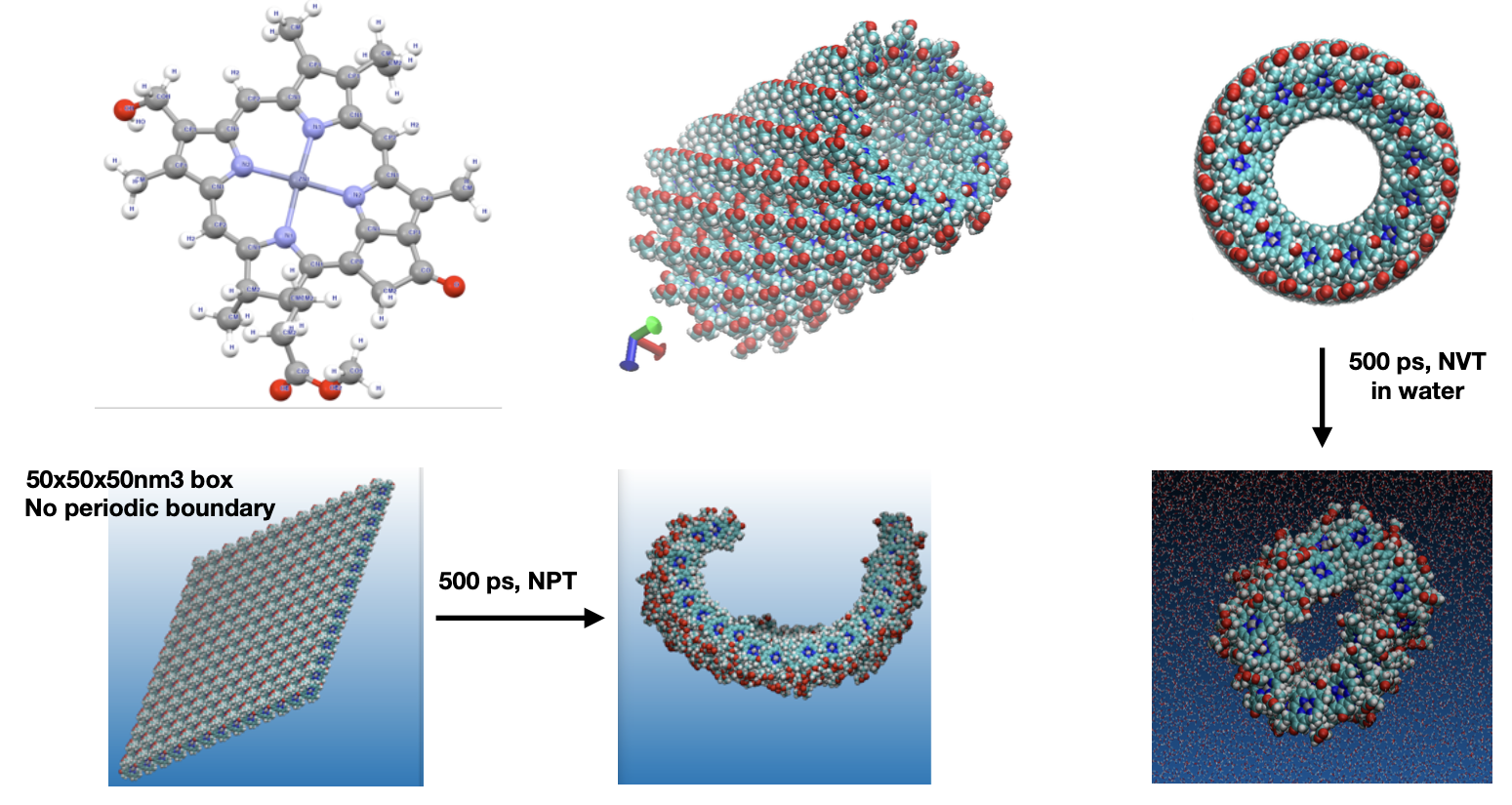
Some photosynthetic bacteria have nano-sized antennas called chlorosomes that capture light efficiently. In this project, I investigate biomimetic zinc-porphyrin supramolecular structures inspired by chlorosomes through atomistic simulations (GROMACS), and perform quantum chemical calculations to understand charge transport properties. These studies aim to understand the structure-function relationships and contribute to the development of new, green, and sustainable solar energy systems. Figure shows the construction and MD studies of the nanotubes.
Structure and Binding Energies of Dimers of Zn(II)-Porphyrin Derivatives
Publication link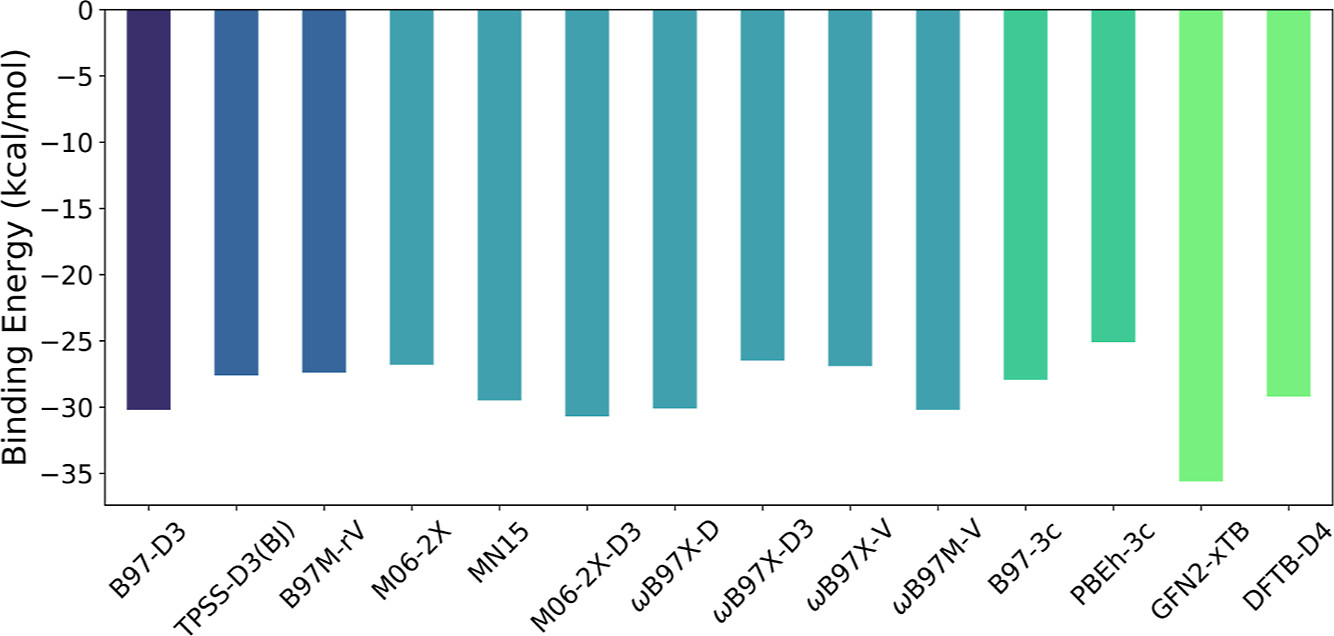
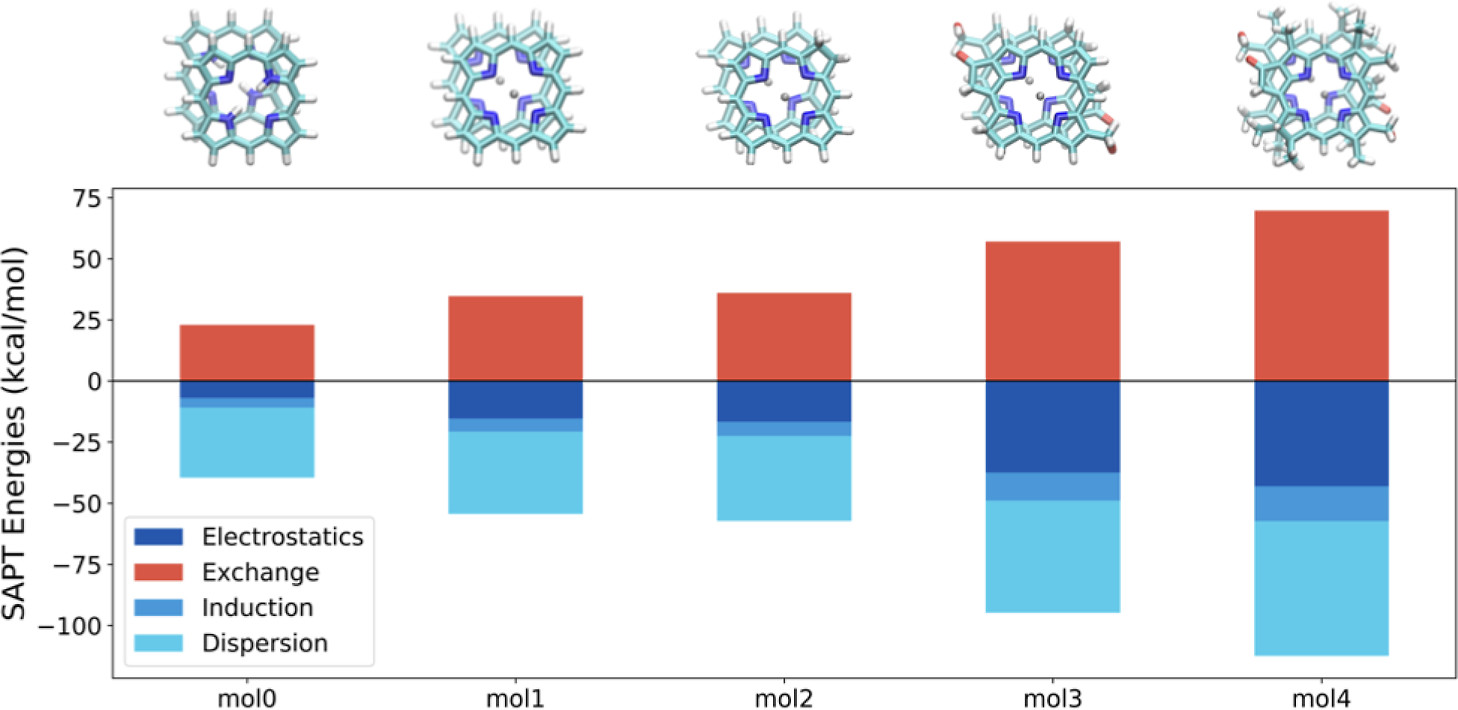
Zinc-complexed porphyrin and chlorophyll derivatives form functional aggregates with remarkable photophysical and optoelectronic properties. Understanding the type and strength of intermolecular interactions between these molecules is essential for designing new materials with desired morphology and functionality. In this project I did a systematic study of the interaction energy (IE) in zinc-porphyrin complexes with increasing structural complexity so that the effect of substitutions on the dimer IEs is quantified. I used semiempirical, density functional, and symmetry-adapted perturbation methods to find a good balance of cost and accuracy (top). Moreover, the types of intermolecular interactions are evaluated using energy decomposition analysis based on the symmetry-adapted perturbation theory (bottom).
A Quantitative structure–property study of reorganization energy
Publication link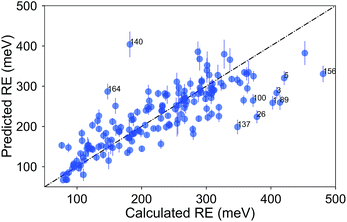
I curated acompound set of 171, which was derived from known p-type OSCs built from moieties such as acenes, thiophenes, and pentalenes and studied the structure-property relationships. It was highlighted in the "Celebrating recent achievements in chemical science in Turkiye" themed collection.
Here is a recent notebook where I explored clustering and interactive plotting with molplotly using this dataset.